Overview 🖼
The objective was to create a molecular dynamics (MD) simulation of Interleukin-1β, a protein largely involved in human inflammation. The protein was observed in a virtual environment meant to mimic human body conditions.
The objective was to create a molecular dynamics (MD) simulation of Interleukin-1β, a protein largely involved in human inflammation. The protein was observed in a virtual environment meant to mimic human body conditions.
The simulation was built using the GROMACS 2024 engine and the CHARMM36m force field. I also used the TIP3P water model (the standard 3-site model). After I processed the initial structure derived from PDB, I placed the protein in a solvated box (a cube-shaped box filled with water molecules) with a 1.0 nm buffer. The system was then neutralized through the addition of sodium and chloride ions. The energy of the system was equilibriated via NVT and NPT to allow it to reach a temperature of 310 Kelvin (human body temperature) and a pressure of 1.0133 bar (1 atm) through a time step of 2 femtoseconds. I used a cut-off scheme of Verlet and PME for electrostatics to make sure all forces are accounted for.
1)
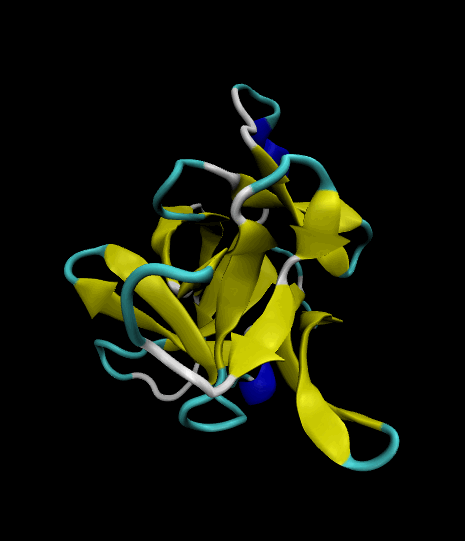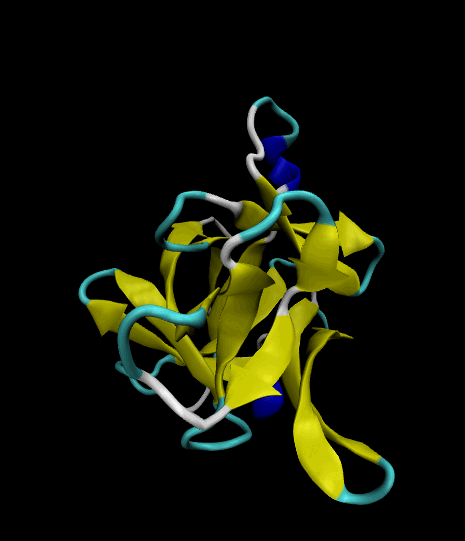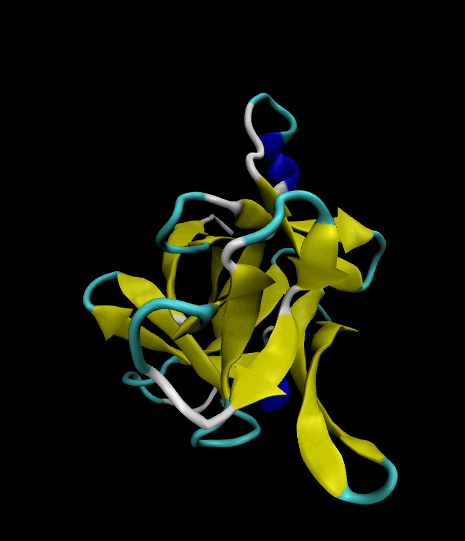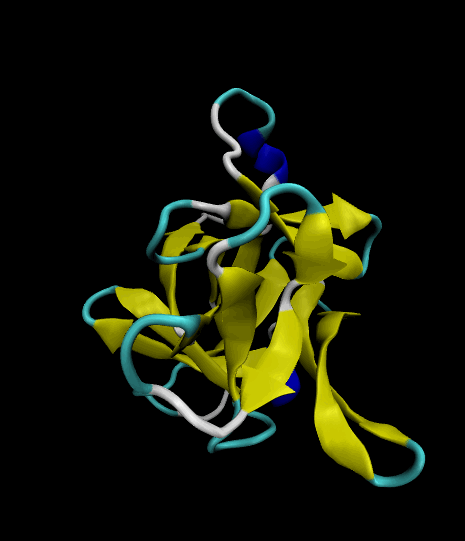
Color-coded by secondary structure.
Styled as a "cartoon."
2)
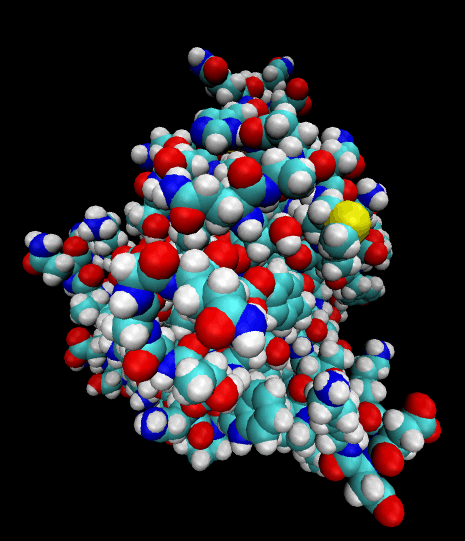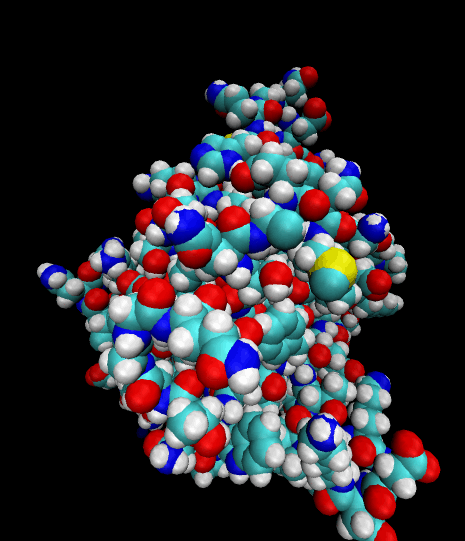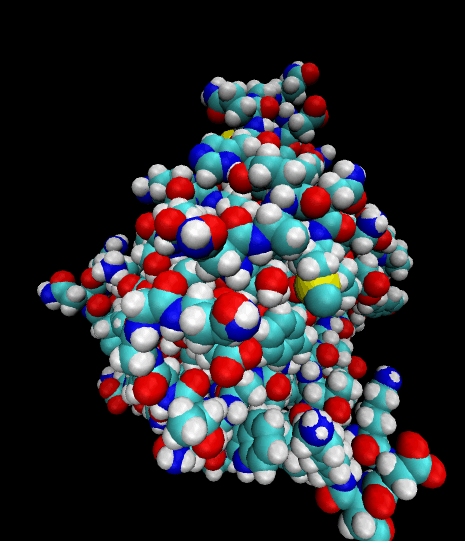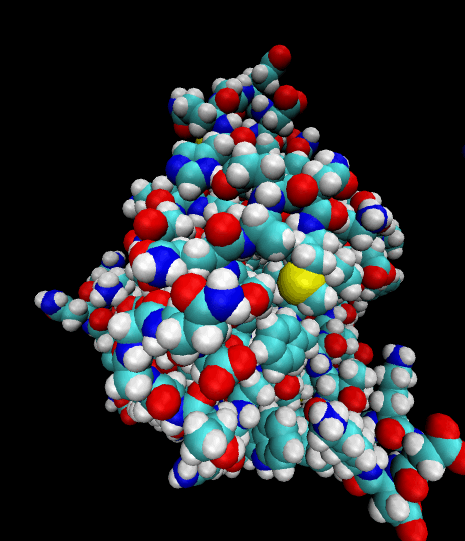
Color-coded by elements.
Styled as an aggregate of atomic bonds.
3)
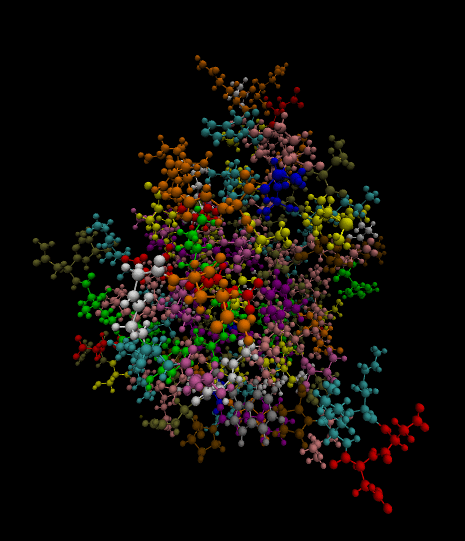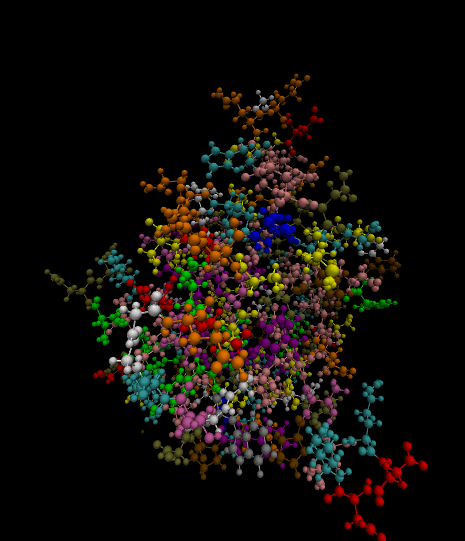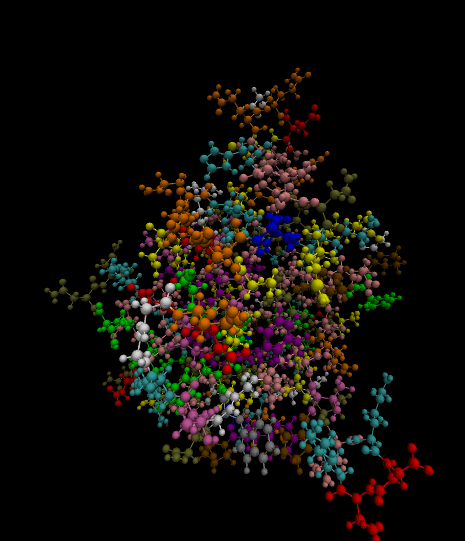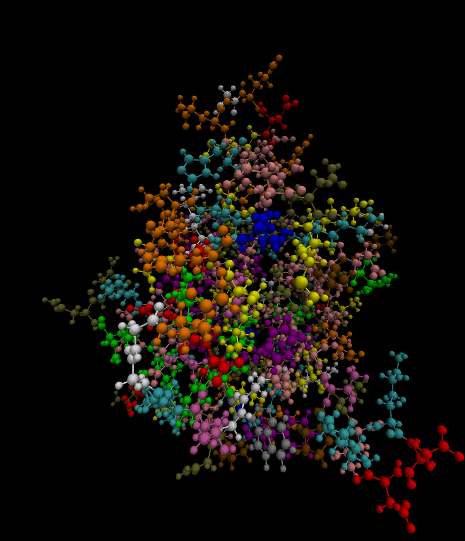
Color-coded by residues present.
Styled as a molecule.
4)
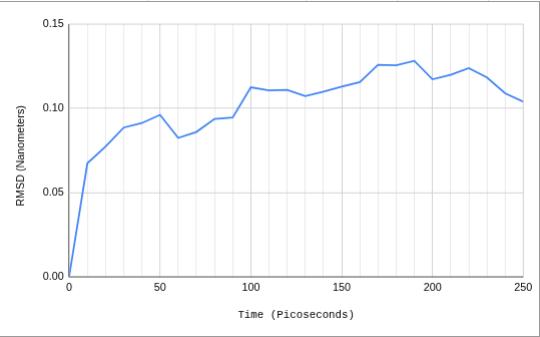
RMSD (Nanometers) plotted against Time (Picoseconds), summarizing the IL-1β protein's movements/displacement during simulation. Plotted curve shows sharp initial rise as protein relaxes from its crystal structure. RMSD oscillates as it plateaus between 0.10 nm and 0.13 nm.
The Root Mean Square Deviation (RMSD) of this simulation reached equilibrium at approximately 0.10 nm within the first 100 picoseconds. This indicates that the protein successfully equilibrated and did not unfold.
The VMD visualizations show that the 12 β strands remained intact. The protein's movement was limited only to surface loops, so the simulation was successful.
Because it maintained the structure of its β-pleated sheets, this could probably be used for other projects in the future.
# generate topology and generate water box
gmx pdb2gmx -f il1b.pdb -o il1b_processed.gro -water tip3p
gmx editconf -f il1b_processed.gro -o il1b_newbox.gro -c -d 1.0 -bt cubic
# solvation
# using ions to neutralize
gmx solvate -cp il1b_newbox.gro -cs spc216.gro -o il1b_solv.gro -p topol.top
gmx genion -s ions.tpr -o il1b_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
gmx grompp -f em.mdp -c il1b_solv_ions.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
# NVT, NPT equilibration
gmx grompp -f nvt.mdp -c em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx mdrun -deffnm npt
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
gmx mdrun -deffnm md_0_1
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_centered.xtc -pbc mol -center
# generate RMSD
gmx rms -s md_0_1.tpr -f md_centered.xtc -o rmsd.xvg -tu nsFeel free to explore the rest of this site.
🔗 Cytokine Home